[Advanced Published online The Keio Journal of Medicine, by J-STAGE]
<Title:> Pachyonychia Congenita: Clinical Features and Future Treatments
<Author(s):> Rebecca L. Mccarthy, Marianne De brito, Edel O’toole
<Corresponding author E-Mill:> r.mccarthy(at)qmul.ac.uk
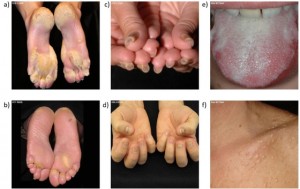
<Abstract:> Pachyonychia congenita (PC) is a rare, autosomal dominant inherited disorder of keratinization that is characterized by a triad of focal palmoplantar keratoderma, plantar pain, and hypertrophic nail dystrophy. It can be debilitating, causing significantly impaired mobility. PC is diagnosed clinically alongside identification of a heterozygous pathogenic mutation in one of five keratin genes: KRT6A, KRT6B, KRT6C, KRT16, or KRT17. Each keratin gene mutation is associated with a distinct clinical phenotype, with variable age of onset and additional features, which has allowed classification by genotype. Additional features include pilosebaceous cysts, follicular hyperkeratosis, natal teeth, oral leukokeratosis, hidradenitis suppurativa, itching, and neurovascular structures. Although classed as rare, the prevalence of PC is likely to be underestimated. There is no cure or specific treatment for PC at present. Current treatments are limited to conservative measures to reduce plantar friction and trauma, mechanical debridement, topical treatments, and treatments for associated features or complications, most commonly infection. However, through active research in collaboration with PC Project, a patient-advocacy group, and the International PC Research Registry, a global registry of PC patients, there are now many new potential therapeutic options on the horizon. This review summarizes the clinical features associated with PC and highlights the current and future treatment of its manifestations.
<Keywords:> pachyonychia congenita, palmoplantar keratoderma, genodermatosis, jadassohn lewandowsky syndrome, jackson lawler syndrome
<URL:> https://www.jstage.jst.go.jp/article/kjm/advpub/0/advpub_2023-0012-IR/_html
<Title:> Pachyonychia Congenita: Clinical Features and Future Treatments
<Author(s):> Rebecca L. Mccarthy, Marianne De brito, Edel O’toole
<Corresponding author E-Mill:> r.mccarthy(at)qmul.ac.uk
<Abstract:> Pachyonychia congenita (PC) is a rare, autosomal dominant inherited disorder of keratinization that is characterized by a triad of focal palmoplantar keratoderma, plantar pain, and hypertrophic nail dystrophy. It can be debilitating, causing significantly impaired mobility. PC is diagnosed clinically alongside identification of a heterozygous pathogenic mutation in one of five keratin genes: KRT6A, KRT6B, KRT6C, KRT16, or KRT17. Each keratin gene mutation is associated with a distinct clinical phenotype, with variable age of onset and additional features, which has allowed classification by genotype. Additional features include pilosebaceous cysts, follicular hyperkeratosis, natal teeth, oral leukokeratosis, hidradenitis suppurativa, itching, and neurovascular structures. Although classed as rare, the prevalence of PC is likely to be underestimated. There is no cure or specific treatment for PC at present. Current treatments are limited to conservative measures to reduce plantar friction and trauma, mechanical debridement, topical treatments, and treatments for associated features or complications, most commonly infection. However, through active research in collaboration with PC Project, a patient-advocacy group, and the International PC Research Registry, a global registry of PC patients, there are now many new potential therapeutic options on the horizon. This review summarizes the clinical features associated with PC and highlights the current and future treatment of its manifestations.
<Keywords:> pachyonychia congenita, palmoplantar keratoderma, genodermatosis, jadassohn lewandowsky syndrome, jackson lawler syndrome
<URL:> https://www.jstage.jst.go.jp/article/kjm/advpub/0/advpub_2023-0012-IR/_html

![Exploration of Microbial Diversity to Discover Novel Molecular Technologies [Published online Keio J Med, 68, 26-26, by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2019/03/68-002-ABST-100x100.jpg)
![Community Pharmacists’ Perceptions and Needs Regarding Oral Healthcare Advice in Japan [Published online in advanced , by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2025/05/2024-0022-OA-100x100.jpg)
![Mechanisms of iPS cell generation and beyond [Published online Keio J Med, 66, 14-14, by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2017/03/66-002-ABST-100x100.jpg)
![Midline Extraperitoneal Approach for Obturator Hernia Repair [Published online in advanced , by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2018/03/2017-0014-OA-100x100.jpg)
![Organoids: Avatars for Personalized Medicine [Published online Keio J Med, 68, 95-95, by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2019/12/68-006-ABST-100x100.jpg)
![The Role of the Adipocyte Hormone Leptin in Alzheimer’s Disease [Published online Keio J Med, 65, 21-21, by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2016/03/65-002-ABST-100x100.jpg)
![RT-PCR Screening Tests for SARS-CoV-2 with Saliva Samples in Asymptomatic People: Strategy to Maintain Social and Economic Activities while Reducing the Risk of Spreading the Virus [Published online in advanced , by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2021/03/2021-0003-OA-100x100.jpg)
![Vitamin D Deficiency with High Intact PTH Levels is More Common in Younger than in Older Women: A Study of Women Aged 39 64 Years [Published online Keio J Med, 65, 33-38, by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2016/06/2015-0010-OA-100x100.jpg)
![Circulating microRNAs: Next-generation Cancer Detection [Published online Keio J Med, 69, 88-96, by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2020/12/2019-0011-OA-100x100.jpg)
![Anatomical Tenodesis Reconstruction Using Free Split Peroneal Brevis Tendon for Severe Chronic Lateral Ankle Instability [Published online Keio J Med, 71, 44-49, by J-STAGE]](http://kjm.pupu.jp/blog/wp-content/uploads/2022/06/2021-0014-OA-100x100.jpg)